
What is LAMA2-CMD?
Laminin Alpha-2–Congenital Muscular Dystrophy (LAMA2-CMD), also known as Merosin-Deficient Congenital Dystrophy Type 1A (MDC1A), is a rare congenital neuromuscular disease that affects 1 to 4 people in every 100,000. The disorder causes weakness and wasting (atrophy) of the muscles used for movement (skeletal muscles). It is the most common form of the congenital muscular dystrophies.
What are the symptoms of LAMA2-CMD?
Babies born with LAMA2-CMD often present poor muscle tone (hypotonia) and muscle weakness at birth, also referred to as “floppy baby syndrome”. These babies may not move as much as other babies and sometimes the first signs are only noticed when the infant has difficulties holding their heads up or sitting up unaided.
Children with LAMA2-CMD reach motor milestones later than other children and only a very small proportion of affected children can walk independently. Muscle weakness does not tend to progress rapidly, and motor function remains relatively stable throughout childhood. When children reach puberty however, and grow taller and heavier, children might experience additional difficulties. Children may also develop or be born with muscle contractures which restrict the movement of their limbs and joints. Most children with LAMA2-CMD also develop a curvature of the spine (scoliosis).
A very common problem in children with LAMA2-CMD is weakness of the respiratory muscles, which results in frequent chest infections and poor breathing at night. Respiratory issues usually stabilise or improve in the first few years. In adolescence however, respiratory function may begin to decline.
Feeding difficulties and the accompanied weight loss are also frequent problems, so it is important to ensure affected children are well nourished. A small number of affected children may experience epileptic seizures. In the worst cases patients may die in childhood.
How is LAMA2-CMD diagnosed?
LAMA2-CMD is usually suspected based on the person’s clinical history and an examination of the symptoms. The specific diagnosis is typically made from studies performed on a muscle biopsy. However, a few additional tests may need to be done.
One of these tests is a blood test, which measures the level of a muscle enzyme called creatine kinase (CK). In individuals with LAMA2-CMD, levels of CK in the blood are typically elevated (often to more than 10 times the normal values).
Brain MRI imaging can be very helpful in identifying LAMA2-CMD as by six months of age, children can display a particular constitution of the white matter in the brain, shown as altered ‘white matter signal’ on brain imaging.
The ultimate diagnosis is obtained by analysing the DNA for changes to the LAMA2 gene. This genetic testing is available at specialised laboratories.
What causes LAMA2-CMD?
LAMA2-CMD is caused by genetic mutations in a gene called Laminin Subunit Alpha-2 (LAMA2). Researchers have identified over 100 mutations in this gene that can cause LAMA2-CMD.
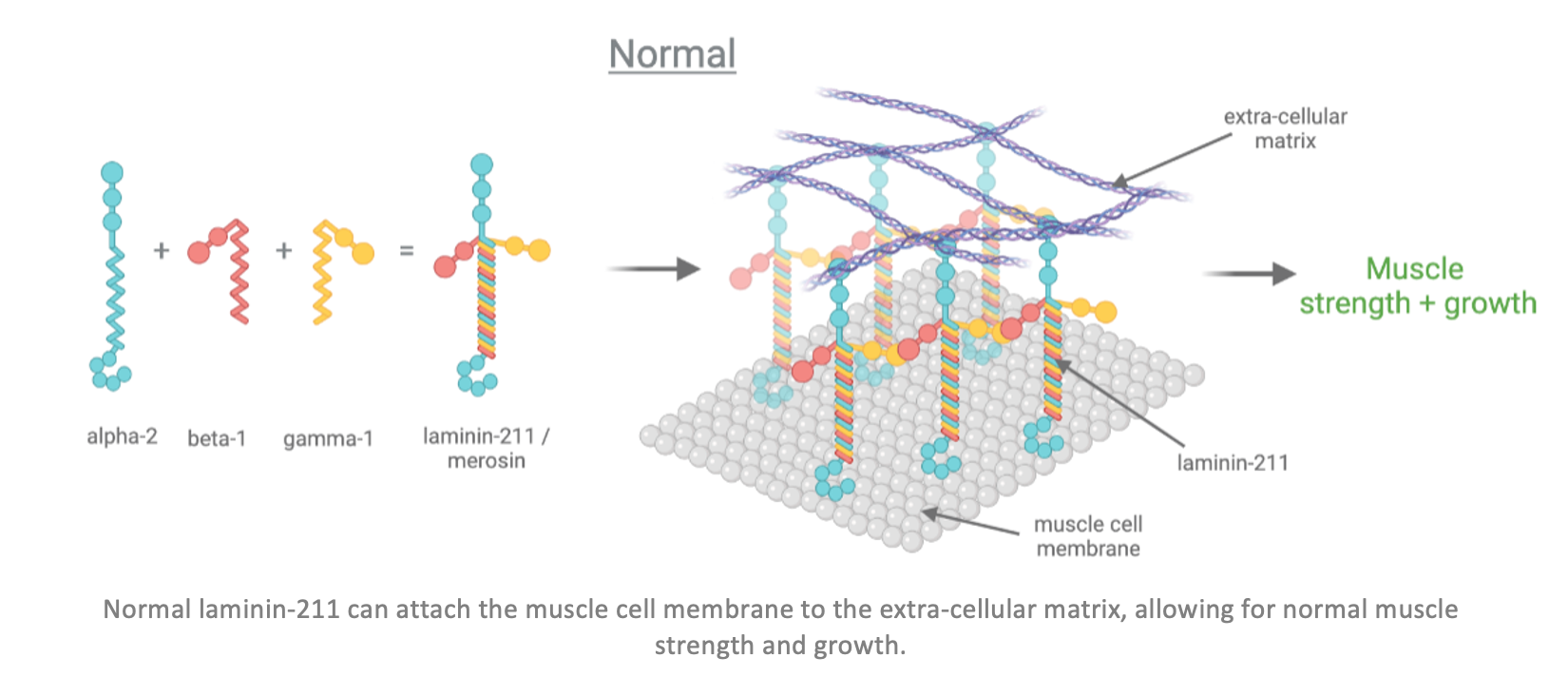
This LAMA2 gene provides cells with instructions to make a part (subunit) of a type of protein called laminins. Laminin proteins are made of three different subunits called alpha, beta, and gamma. The LAMA2 gene provides instructions for the alpha-2 subunit.
This subunit, together with the beta-1 and gamma-1 subunits, forms the laminin-211 protein, which is also called merosin. This explains why the disease is sometimes called MDC1 or LAMA2-CMD.
In the human body, muscle cells and muscle fibres are surrounded by and connected to each other by a kind of web called the extra-cellular matrix. Laminin-211 plays an important role in the muscles used for movement (skeletal muscles), as it connects and attaches the membranes of the muscle cells to the extracellular matrix. By connecting the muscle cells to the extra-cellular matrix, laminins help regulate cell growth, cell movement and cell adhesion which gives the muscles strength and stability and allows them to grow.
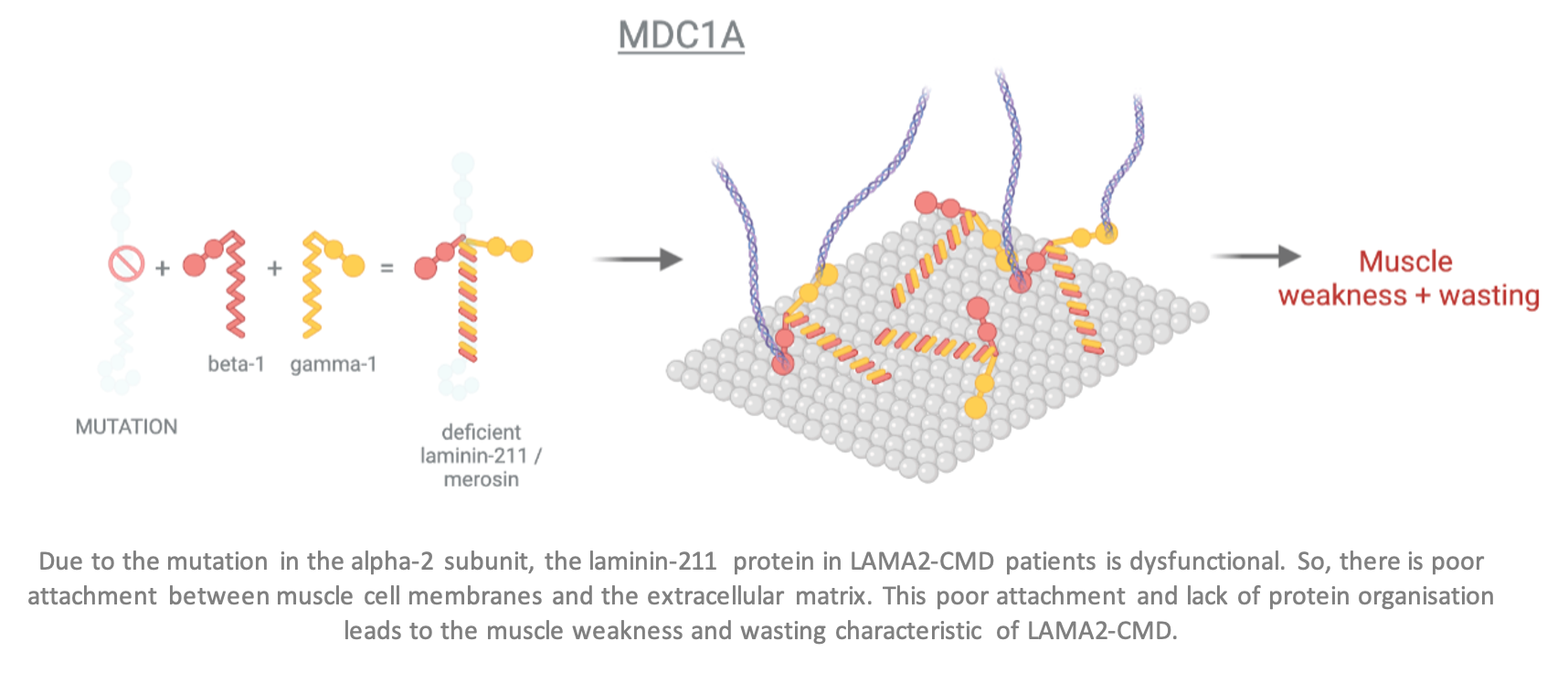
In LAMA2-CMD, laminin-211 is dysfunctional due to the mutation in the LAMA2 gene. This means the muscle cells cannot attach to each other via the extracellular matrix, resulting in reduced muscle strength and stability which causes the signs and symptoms of LAMA2-CMD.
In cases where LAMA2 is completely absent, LAMA2-CMD generally appears as a more severe, early-onset form. When LAMA2 is only partially reduced, the condition can be milder and have a late-onset, sometimes only detected during adulthood.
What are the current treatments for LAMA2-CMD?
There is currently no available cure for LAMA2-CMD but there are existing treatments and therapies to help alleviate the symptoms. Physiotherapy can help prevent muscle contractures, and a specific exercise programme should be led by a physiotherapist as early as possible. Often, surgery may be required to help release the contractures.
It is particularly important to monitor respiratory function during the night and children may need to use a ventilator at night with a special facial or nasal mask to assist them breathing. For some patients this is insufficient, and a tracheostomy procedure is needed. Respiratory issues usually stabilise or improve in the first few years, but if respiratory function starts to decline during adolescence, further ventilator support may be required.
For the minority of children with LAMA2-CMD that present seizures, these can usually be well controlled with anti-epileptic medication. It is also important to make sure that children with LAMA2-CMD are well nourished. Often the swallowing muscles are weak and feeding supplements are needed. In some cases, swallowing problems can occur since the muscles responsible for swallowing may be weak. To address this, a small surgical procedure, called a gastrostomy can be performed.
Children and adults with LAMA2-CMD should ideally be followed regularly in a specialist neuromuscular clinic, with access to physiotherapy, orthotic, respiratory, orthopaedic, spinal and genetic specialists as needed.
What new therapies are currently being researched?
A lot has been learned about LAMA2-CMD by scientists in recent years and there is growing evidence on several new therapeutic approaches for LAMA2-CMD.
One approach is autologous stem cell therapy. Here, muscle stem cells are withdrawn from patients themselves and the mutated gene is replaced with a healthy version in the lab. The corrected muscle stem cells are then returned to the patients so that they can generate new healthy muscle and counteract the muscle weakness for an extended period of time. Such an approach is currently being investigated in a Phase I/II clinical trial by a new European research collaboration.
A second approach is gene therapy. Here, a replacement version of the LAMA2 gene, or other replacement proteins can be introduced directly into the patient’s muscle cells, allowing them to grow and function more normally.
Another approach is using drug treatments that buffer the muscle loss caused by genetic mutations. Protein replacement therapy is also being developed and will undergo testing clinical trials in the future. These treatments are also an option that are currently being explored and could be combined with other therapies.
While these options are promising, more research and communication between experts, clinicians and biotechnology companies is needed to improve their efficacy and accelerate their implementation into the standard of care for LAMA2-CMD.
As always, if you are in the need for any information or are looking to contact an LAMA2-CMD expert, we invite you to contact us directly here.
Support the research for a cure and donate now!
The driving forces behind Lama2.com
Bekijk alle partners

